Computational Biology

Education
University of Texas at Dallas, Richardson, TX
Master of Science, Computer Science, GPA 4.0/4.0, Recipient of Certificate of Academic Excellence
Computational Skills
- Computer Science
- General-purpose Programming Languages: Java, Python, Shell Scripting
- Databases: MySQL, MongoDB, Oracle SQL, PL/SQL
- Web Programming (Front-End): HTML/HTML5, CSS3, Bootstrap, JavaScript, JQuery, AJAX, XML, JSON
- Web Programming (Back-End): Spring MVC, Servlet, JSP, Hibernate, PHP
- Web Programming (Web Services): Spring REST, Spring Boot
- Software Tools/Technologies: AWS, Eclipse, IntelliJ, SQL Developer, Apache Tomcat, Adobe Illustrator, Git/GitHub, Postman
- Operating Systems: Windows, Linux
- Data Science
- Programming Languages: Python, R, Spark, MATLAB
- Software Tools/Libraries: SciKit-Learn (GitHub), NumPy, SciPy, Pandas, Matplotlib, TensorFlow, Keras, PyTorch, OpenCV
- Big Data Technologies: Hadoop, Spark
- Machine Learning/Artificial Intelligence (AI) Algorithmns (GitHub): Decision Tree/Random Forest, Artificial Neural Networks (ANN), Naive Bayes, K-Nearest Neighbors (KNN), Linear Regression, Logistic Regression, Support Vector Machines (SVM), K-means Clustering, Gaussian Mixture Model (GMM) Clustering, AdaBoost, Cross-validation, Deep Learning, Blind/Informed Search, Minimax Algorithm, Alpha-Beta Pruning, Q-Learning
- Graphical Statistical Algorithms (GitHub): Bayesian Network, Hidden Markov Model (HMM), Belief Propagation, Gibbs Sampling, Expectation‐Maximization (EM) Algorithm, Structure Learning
- Computer Vision Algorithms: Histogram Equalization, Haar Cascade Object Detection (GitHub)
- Bioinformatics
- Amplicon NGS (Next Generation Sequencing) (ref)
- RNA sequencing/RNA-seq (HISAT2, Samtools, StringTie, DESeq2, Ballgown)
- Whole exome sequencing/WES (FastQC, BWA, Picard, Samtools, VarScan 2, GATK Mutect2, ANNOVAR, IGV)
- Fusion transcripts detection (STAR, Arriba)
- Cell segmentation (Cellpose, TrackMate, ImageJ/Fiji)
Current Projects
Comparisons of cellular segmentation algorithms for mammalian cells
We will use CRISPR/SpCas9 technology to create mammalian cell line models which stably express MKATE red fluorescence proteins on plasma membranes. Next, different deep learning-based cellular segmentation algorithms (e.g., Cellpose-TrackMate, PixelLib) will be evaluated using fluorescence microscopy images acquired from the MKATE stable cells.
Unraveling mechanisms of exceptional response to cyclin-dependent kinase (CDK) inhibitors in patients with metastatic breast cancer
We will use whole exome sequencing (WES) assay to identify potential somatic or germline single-nucleotide variations (SNVs) which contribute to the exceptional response to CDK inhibitors in HR-positive, HER2-negative breast cancer patients.
Publications
Machine learning-based approaches for identifying human blood cells harboring CRISPR-mediated fetal chromatin domain ablations
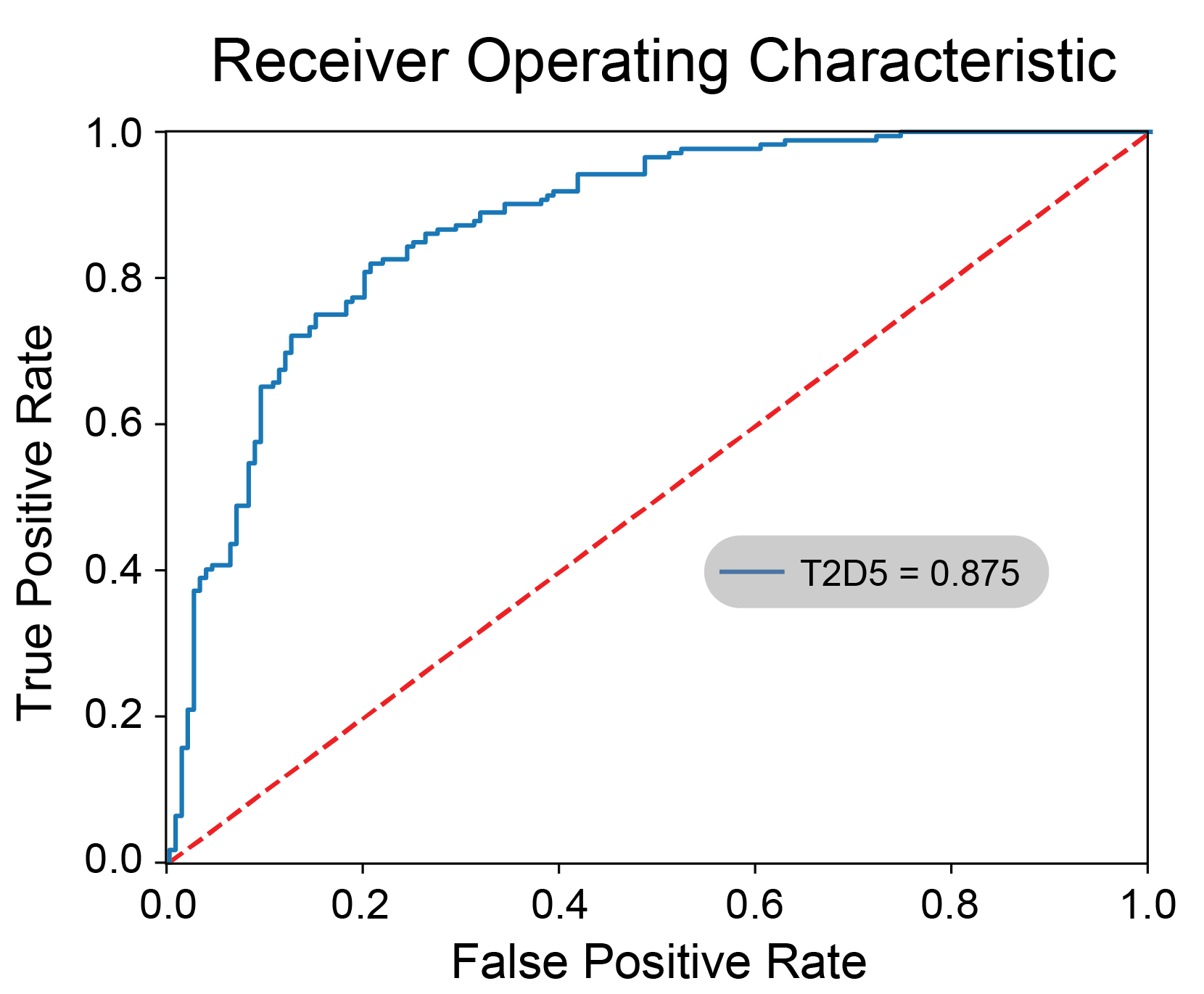
Two common hemoglobinopathies, sickle cell disease (SCD) and β-thalassemia, arise from genetic mutations within the β-globin gene. In this work, we identified a 500-bp motif (Fetal Chromatin Domain, FCD) upstream of human γ-globin locus and showed that the removal of this motif using CRISPR technology reactivates the expression of γ-globin. Next, we present two different cell morphology-based machine learning approaches that can be used identify human blood cells (KU-812) that harbor CRISPR-mediated FCD genetic modifications. Three candidate models from the first approach, which uses multilayer perceptron algorithm (MLP 20-26, MLP26-18, and MLP 30-26) and flow cytometry-derived cellular data, yielded 0.83 precision, 0.80 recall, 0.82 accuracy, and 0.90 area under the ROC (receiver operating characteristic) curve when predicting the edited cells. In comparison, the candidate model from the second approach, which uses deep learning (T2D5) and DIC microscopy-derived imaging data, performed with less accuracy (0.80) and ROC AUC (0.87). We envision that equivalent machine learning-based models can complement currently available genotyping protocols for specific genetic modifications which result in morphological changes in human cells. download
Cell morphology-based machine learning models for human cell state classification
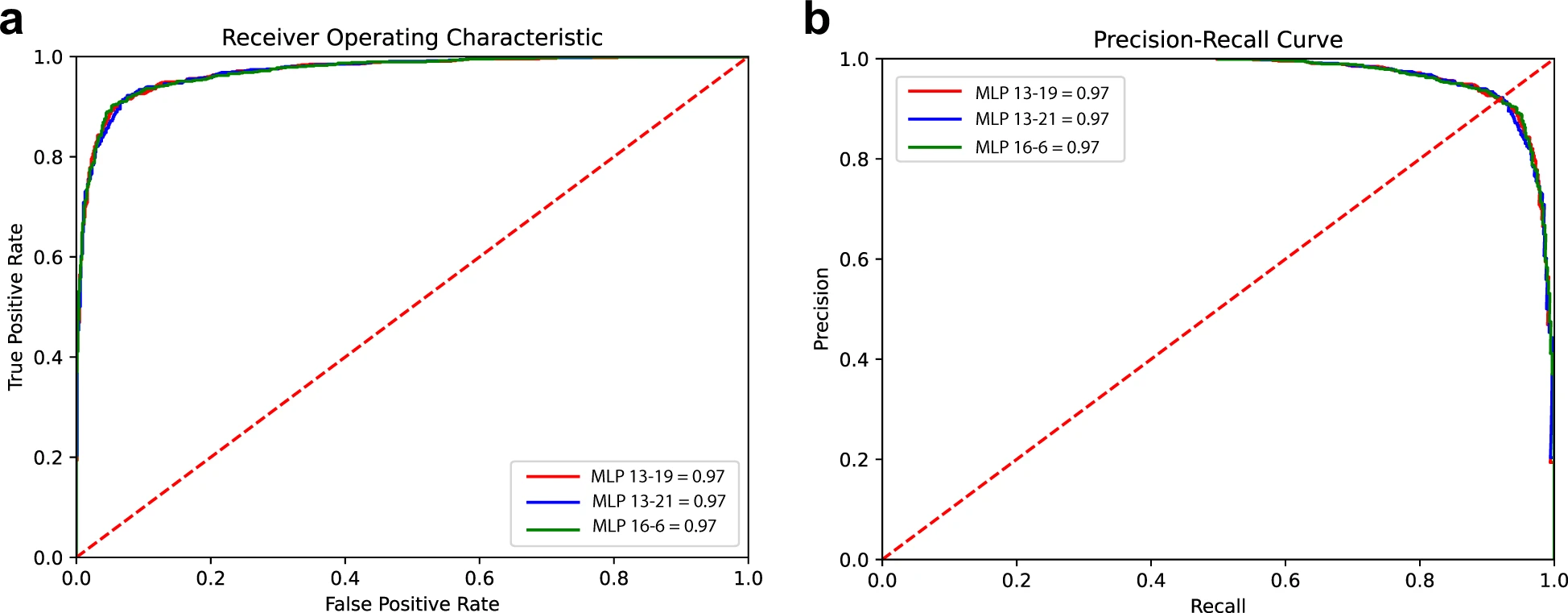
Herein, we implement and access machine learning architectures to ascertain models that differentiate healthy from apoptotic cells using exclusively forward (FSC) and side (SSC) scatter flow cytometry information. To generate training data, colorectal cancer HCT116 cells were subjected to miR-34a treatment and then classified using a conventional Annexin V/propidium iodide (PI)-staining assay. The apoptotic cells were defined as Annexin V-positive cells, which include early- and late-apoptotic cells, necrotic cells, as well as other dying or dead cells. In addition to fluorescent signal, we collected cell size and granularity information from the FSC and SSC parameters. Both parameters are subdivided into area, height, and width, thus providing a total of six numerical features that informed and trained our models. A collection of logistical regression, random forest, k-nearest neighbor, multilayer perceptron, and support vector machine were trained and tested for classification performance in predicting cell states using only the six aforementioned numerical features. Out of 1,046 candidate models, a multilayer perceptron was chosen with 0.91 live precision, 0.93 live recall, 0.92 live f-value and 0.97 live area under the ROC curve when applied on standardized data. We discuss and highlight differences in classifier performance and compare the results to the standard practice of forward and side scatter gating, typically performed to select cells based on size and/or complexity. We demonstrate that our model, a ready-to-use module for any flow cytometry-based analysis, can provide automated, reliable, and stain-free classification of healthy and apoptotic cells using exclusively size and granularity information. download
Coevolutionary couplings unravel PAM-Proximal constraints of CRISPR-SpCas9
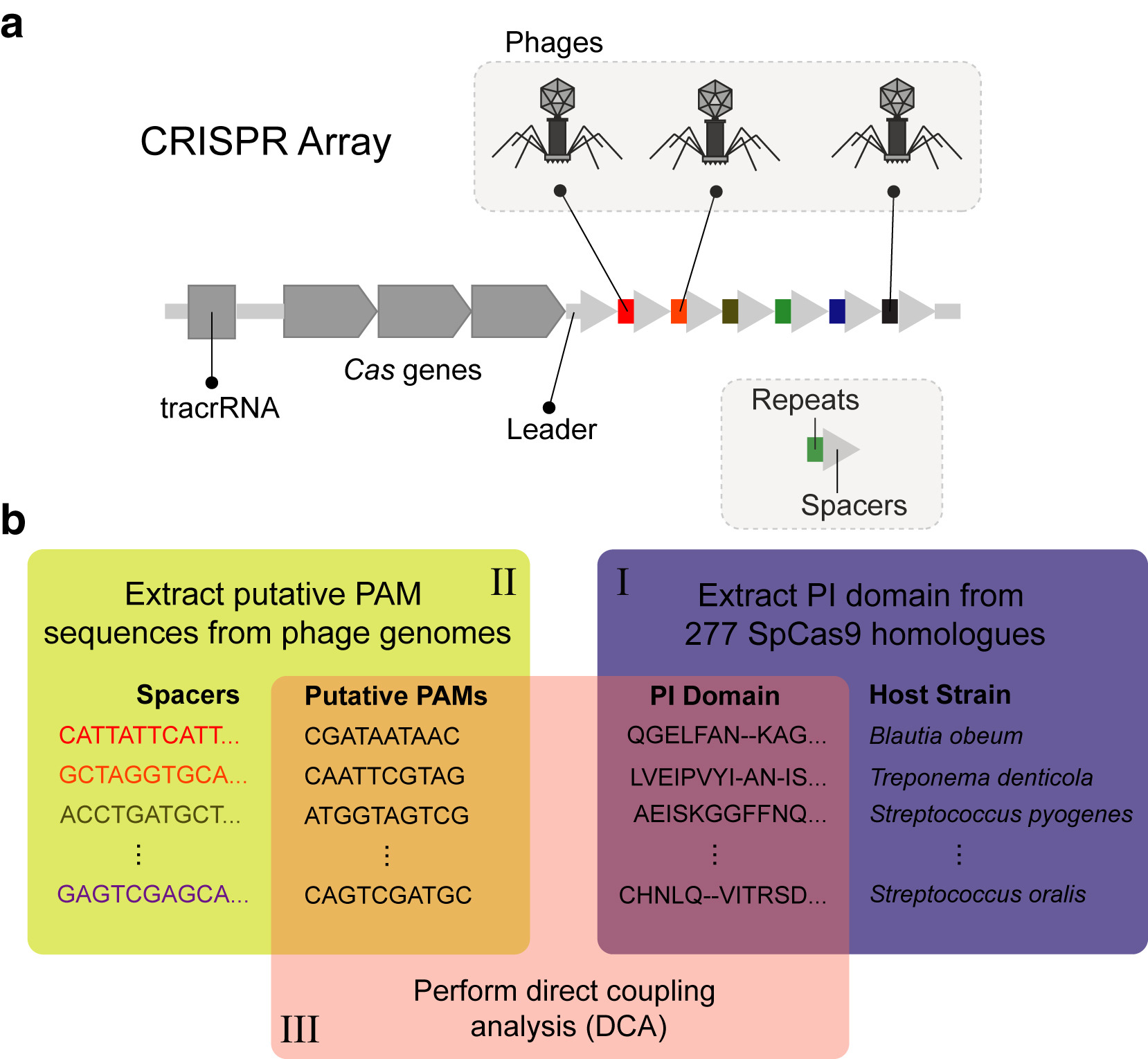
The clustered regularly interspaced short palindromic repeats (CRISPR) system, an immune system analog found in prokaryotes, allows a single-guide RNA to direct a CRISPR-associated protein (Cas) with combined helicase and nuclease activity to DNA. The presence of a specific protospacer adjacent motif (PAM) next to the DNA target site plays a crucial role in determining both efficacy and specificity of gene editing. Herein, we introduce a coevolutionary framework to computationally unveil nonobvious molecular interactions in CRISPR systems and experimentally probe their functional role. Specifically, we use direct coupling analysis, a statistical inference framework used to infer direct coevolutionary couplings, in the context of protein/nucleic acid interactions. Applied to Streptococcus pyogenes Cas9, a Hamiltonian metric obtained from coevolutionary relationships reveals, to our knowledge, novel PAM-proximal nucleotide preferences at the seventh position of S. pyogenes Cas9 PAM (5'-NGRNNNT-3'), which was experimentally confirmed by in vitro and functional assays in human cells. We show that coevolved and conserved interactions point to specific clues toward rationally engineering new generations of Cas9 systems and may eventually help decipher the diversity of this family of proteins. download
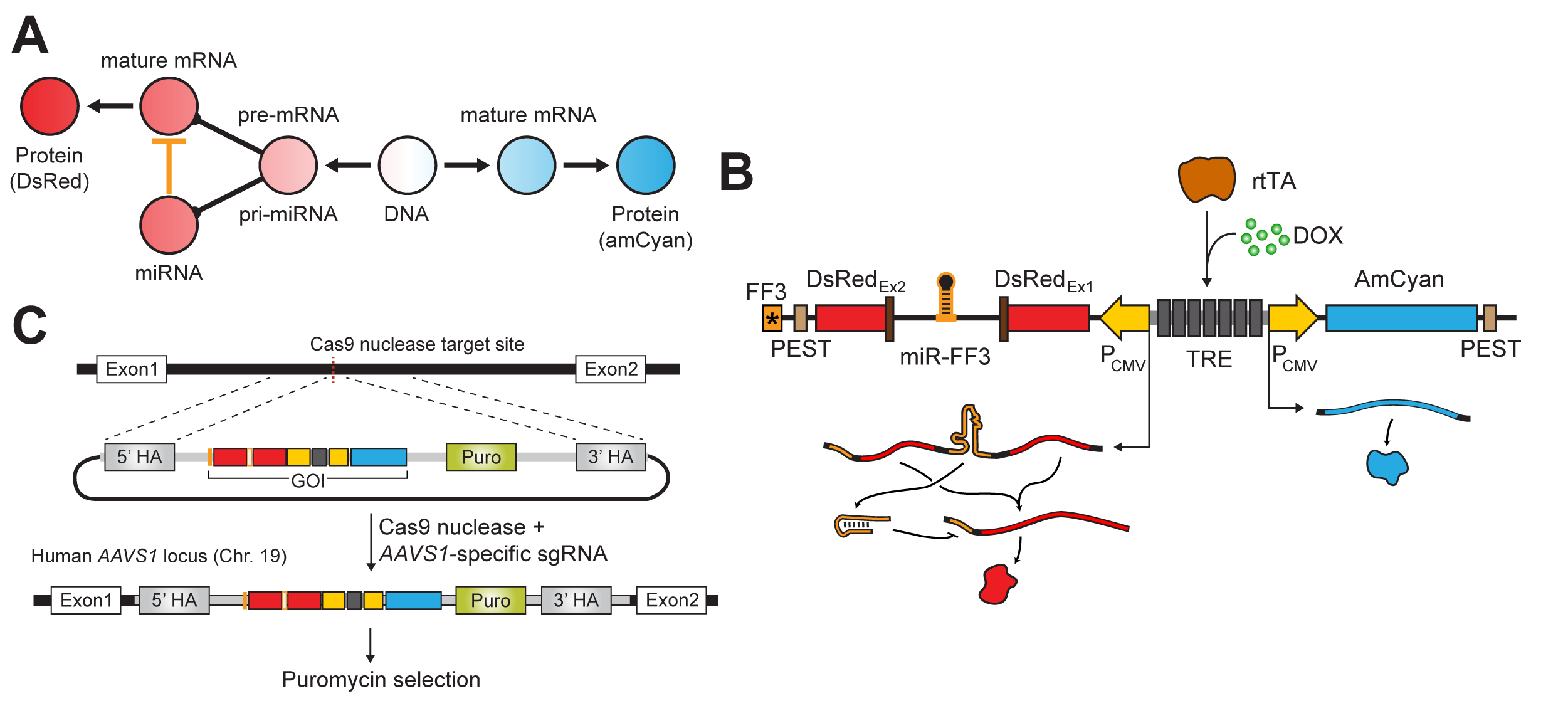
Uncoupling gene expression noise along the central dogma using genome engineered human cell lines
Eukaryotic protein synthesis is an inherently stochastic process. This stochasticity stems not only from variations in cell content between cells but also from thermodynamic fluctuations in a single cell. Ultimately, these inherently stochastic processes manifest as noise in gene expression, where even genetically identical cells in the same environment exhibit variation in their protein abundances. In order to elucidate the underlying sources that contribute to gene expression noise, we quantify the contribution of each step within the process of protein synthesis along the central dogma. We uncouple gene expression at the transcriptional, translational, and post-translational level using custom engineered circuits stably integrated in human cells using CRISPR. We provide a generalized framework to approximate intrinsic and extrinsic noise in a population of cells expressing an unbalanced two-reporter system. Our decomposition shows that the majority of intrinsic fluctuations stem from transcription and that coupling the two genes along the central dogma forces the fluctuations to propagate and accumulate along the same path, resulting in increased observed global correlation between the products. download
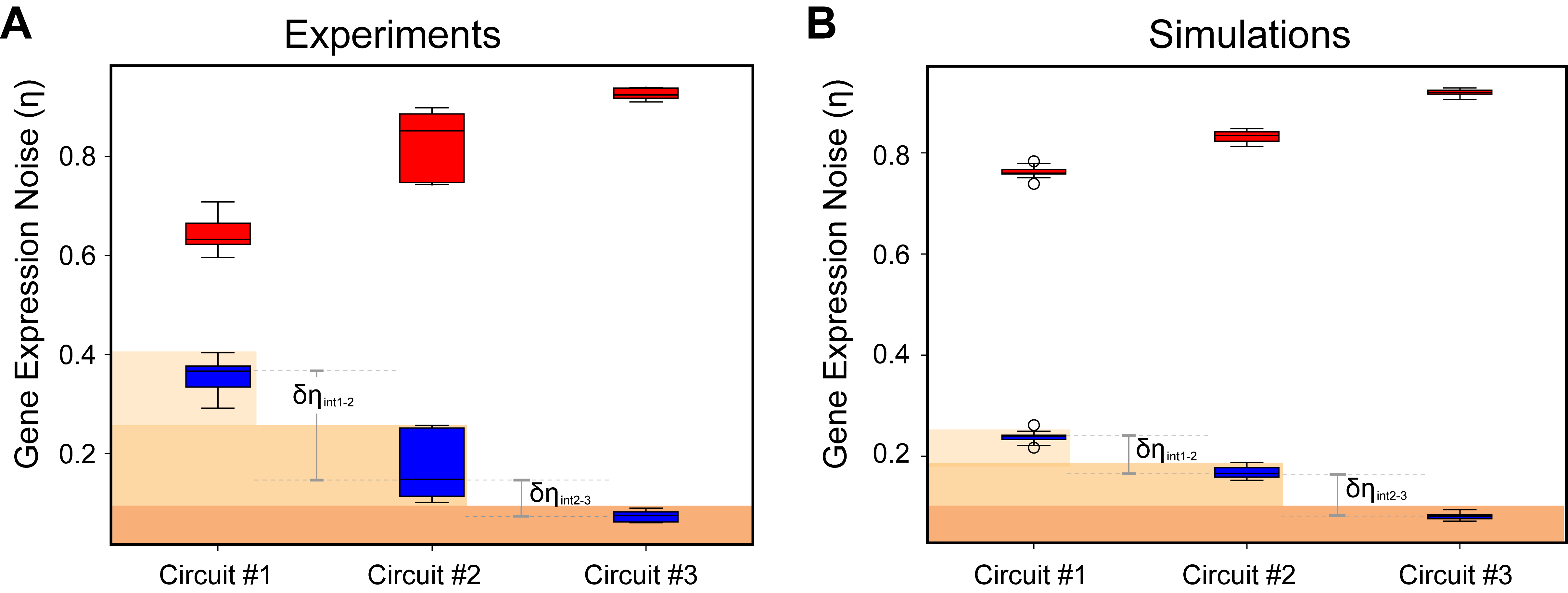
Robust filtering and noise suppression in intragenic miRNA-mediated host regulation
MicroRNAs (miRNAs) are short non-coding RNA molecules that regulate gene expression post-transcriptionally by binding to target messenger RNAs (mRNAs). Many human miRNAs are intragenic, located within introns of protein-coding sequence (host). Intriguingly, a percentage of intragenic miRNAs downregulate the host transcript forming an incoherent feedforward motif topology. Here, we study intragenic miRNA-mediated host gene regulation using a synthetic gene circuit stably integrated within a safe harbor locus of human cells. When the intragenic miRNA is directed to inhibit the host transcript, we observe a reduction in reporter expression accompanied by output filtering and noise reduction. Specifically, the system operates as a filter with respect to promoter strength, with the threshold being robust to promoter strength and measurement time. Additionally, the intragenic miRNA regulation reduces expression noise compared to splicing-alone architecture. Our results provide a new insight to miRNA-mediated gene expression, with direct implications to gene therapy and synthetic biology applications. download
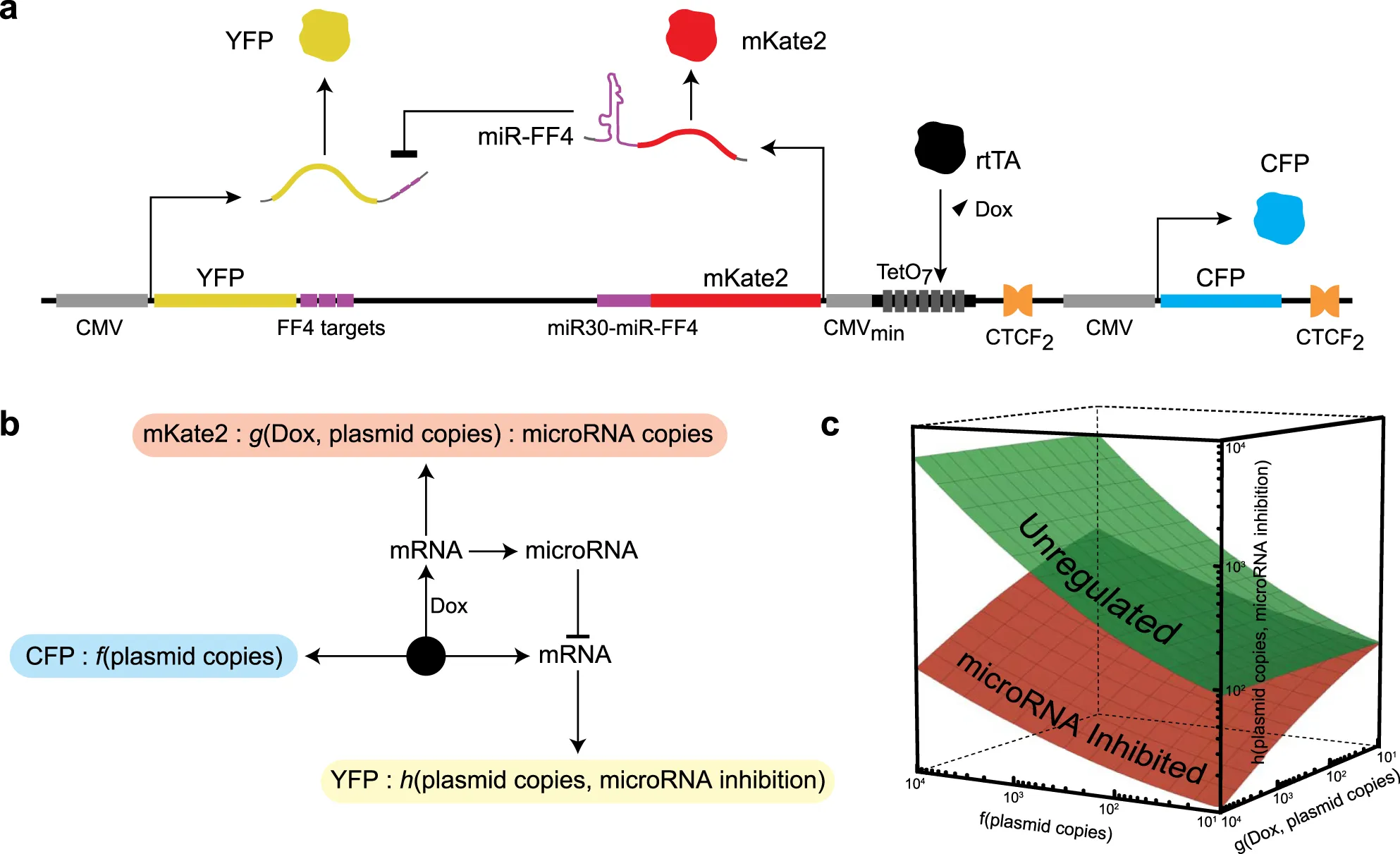
Mapping the operational landscape of microRNAs in synthetic gene circuits
MicroRNAs are a class of short, noncoding RNAs that are ubiquitous modulators of gene expression, with roles in development, homeostasis, and disease. Engineered microRNAs are now frequently used as regulatory modules in synthetic biology. Moreover, synthetic gene circuits equipped with engineered microRNA targets with perfect complementarity to endogenous microRNAs establish an interface with the endogenous milieu at the single-cell level. The function of engineered microRNAs and sensor systems is typically optimized through extensive trial-and-error. Here, using a combination of synthetic biology experimentation in human embryonic kidney cells and quantitative analysis, we investigate the relationship between input genetic template abundance, microRNA concentration, and output under microRNA control. We provide a framework that employs the complete operational landscape of a synthetic gene circuit and enables the stepwise development of mathematical models. We derive a phenomenological model that recapitulates experimentally observed nonlinearities and contains features that provide insight into the microRNA function at various abundances. Our work facilitates the characterization and engineering of multi-component genetic circuits and specifically points to new insights on the operation of microRNAs as mediators of endogenous information and regulators of gene expression in synthetic biology. download
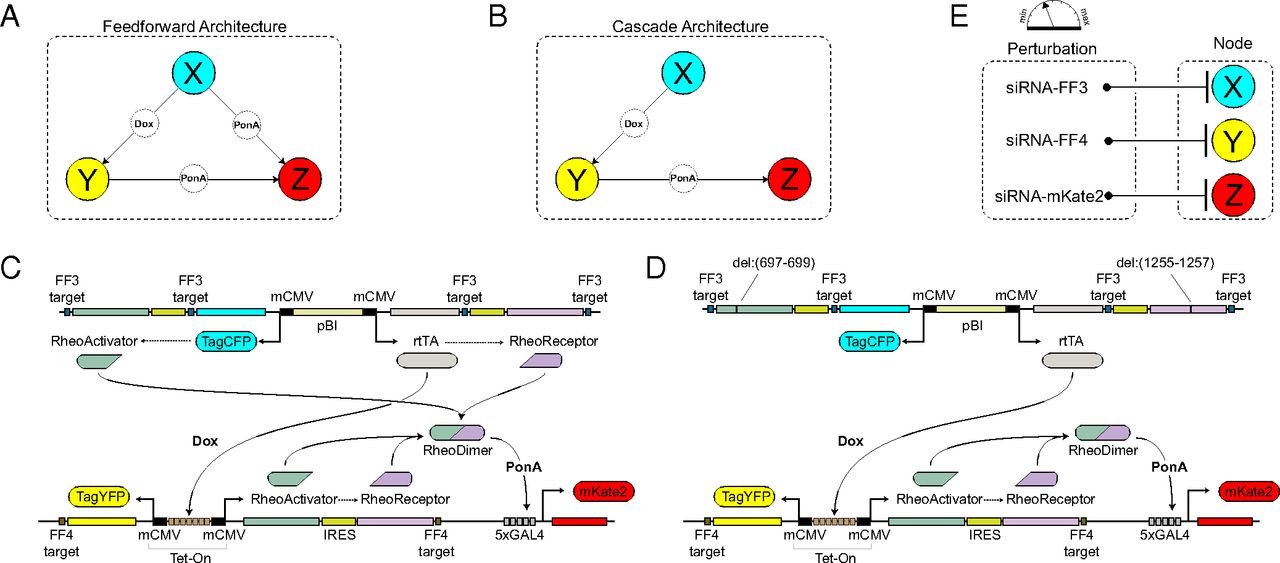
Discriminating direct and indirect connectivities in biological networks
Reverse engineering of
biological pathways involves an iterative process between experiments, data processing, and theoretical analysis.
Despite concurrent advances in quality and quantity of data as well as computing resources and algorithms,
difficulties in deciphering direct and indirect network connections are prevalent. Here, we adopt the notions of
abstraction, emulation, benchmarking, and validation in the context of discovering features specific to this family
of connectivities. After subjecting benchmark synthetic circuits to perturbations, we inferred the network connections
using a combination of nonparametric single-cell data resampling and modular response analysis. Intriguingly, we
discovered that recovered weights of specific network edges undergo divergent shifts under differential perturbations,
and that the particular behavior is markedly different between topologies. Our results point to a conceptual advance
for reverse engineering beyond weight inference. Investigating topological changes under differential perturbations
may address the longstanding problem of discriminating direct and indirect connectivities in biological networks.
download
CRISPR-based self-cleaving mechanism for controllable gene delivery in human cells
Controllable gene
delivery via vector-based systems remains a formidable challenge in mammalian synthetic biology and a desirable
asset in gene therapy applications. Here, we introduce a methodology to control the copies and residence time of a
gene product delivered in host human cells but also selectively disrupt fragments of the delivery vehicle. A
crucial element of the proposed system is the CRISPR protein Cas9. Upon delivery, Cas9 guided by a custom RNA
sequence cleaves the delivery vector at strategically placed targets thereby inactivating a co-expressed gene of
interest. Importantly, using experiments in human embryonic kidney cells, we show that specific parameters of the
system can be adjusted to fine-tune the delivery properties. We envision future applications in complex synthetic
biology architectures, gene therapy and trace-free delivery.
download

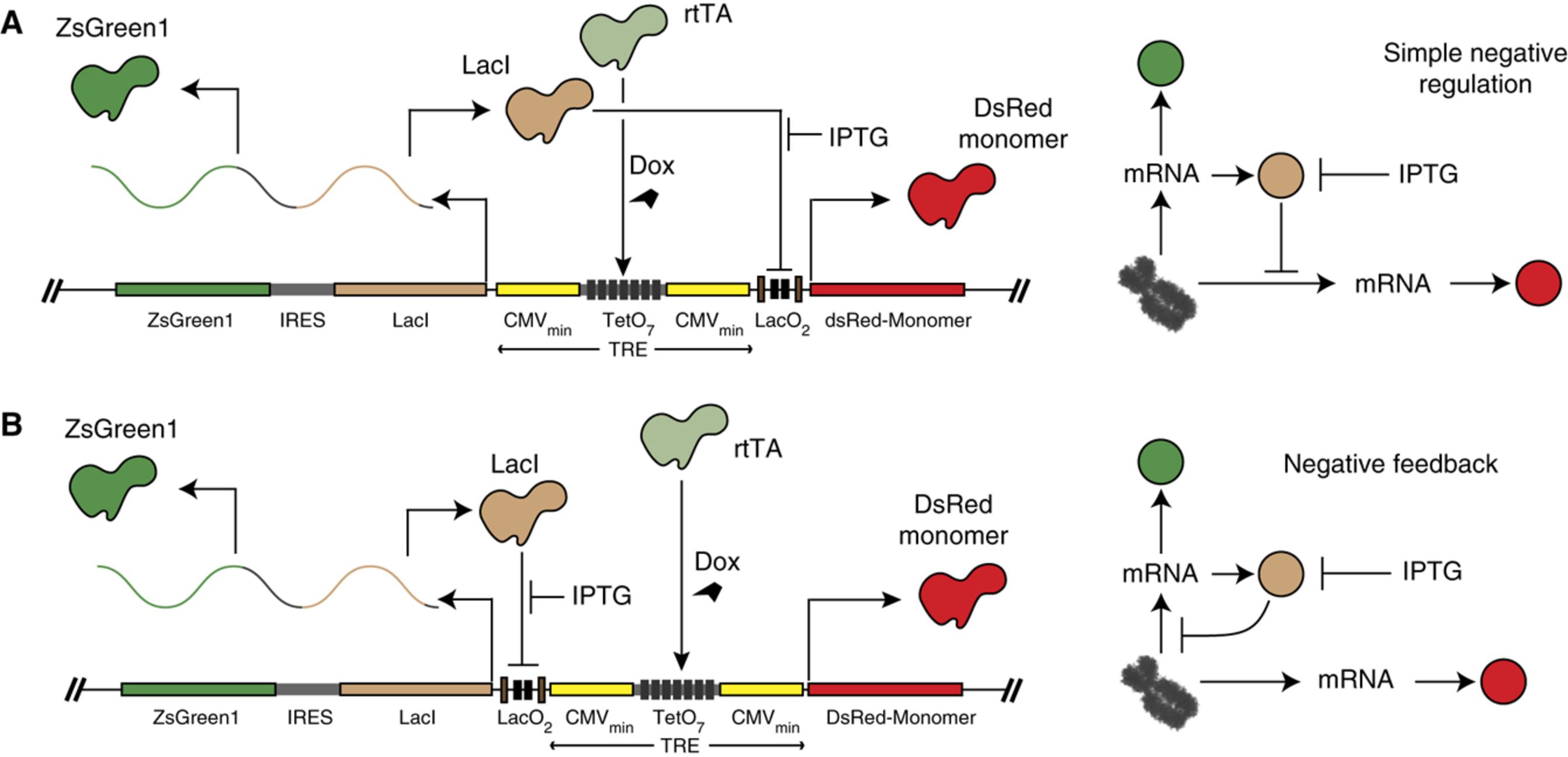
Synthetic mammalian transgene negative autoregulation
Biological networks contain overrepresented small-scale topologies, typically called motifs. A frequently appearing motif is the transcriptional negative-feedback loop, where a gene product represses its own transcription. Here, using synthetic circuits stably integrated in human kidney cells, we study the effect of negative-feedback regulation on cell-wide (extrinsic) and gene-specific (intrinsic) sources of uncertainty. We develop a theoretical approach to extract the two noise components from experiments and show that negative feedback results in significant total noise reduction by reducing extrinsic noise while marginally increasing intrinsic noise. We compare the results to simple negative regulation, where a constitutively transcribed transcription factor represses a reporter protein. We observe that the control architecture also reduces the extrinsic noise but results in substantially higher intrinsic fluctuations. We conclude that negative feedback is the most efficient way to mitigate the effects of extrinsic fluctuations by a sole regulatory wiring. download